
{kind=link}
So long, Japan, and thanks for all the fish
July 23, 2018
My visit to Japan began with a one-week stay at Osaka University with Dr. Chizu Kato. Here, I learned about her research. Specifically, about the experimental and theoretical determination of zinc and gallium isotopic fractionation in lunar samples. I was also treated to presentations by several graduate students in their research group who were happy to practice their English because at Osaka University, graduate students aren’t given the luxury of defending their thesis in their mother tongue. It was a really neat opportunity to get exposed to some different applications of isotope geochemistry. Their research tended to focus on the separation nuclear fission products prior to long-term storage. I also gave a short presentation on some of the research I’ve been working on so far. Thankfully, I didn’t have to give it in Japanese.

Theoretical chemistry research group at Tokyo Metropolitan University.
I traveled to Tokyo Metropolitan University on June 25th to work with Dr. Minori Abe and spent my first week there getting familiar with Gaussian, a computational chemistry software package that was initially developed in 1970 by John Pople and his research group at Carnegie Mellon University. Pople received the Nobel prize in Chemistry in 1998 for his contributions to the field of computational chemistry. Interestingly, his association with Gaussian ended in 1991 after being banned for modifying the software (the Gaussian user agreement seems quite controversial and there’s a whole website devoted to those who have been banned from using the software for performing competing research: http://www.bannedbygaussian.org/). The whole ordeal is likely about as dramatic as the scientific (or at least theoretical chemistry) community gets.
Gaussian is the software that I primarily used while in Japan, although a portion of my time was devoted to learning General Atomic and Molecular Electronic Structure System (GAMESS), a free software that can perform similar functions to Gaussian. I decided to gain familiarity with both in case I wasn’t able to access Gaussian once back at Queen’s University. Fortunately, now that I’m back, I’ve found I’ll be able to get access to Gaussian through our computing department.
I approached learning Gaussian by recreating some of the results in a paper Dr. Abe and Dr. Toshiyuki Fujii worked on together (Fujii et al., 2013) related to copper isotope fractionation between aqueous compounds relevant to low temperature geochemistry and biology. Once I was able to produce comparable results, I moved on to metal-amino acid complexes computed in Fujii et al. (2014) before dabbling with copper complexes within amyloid beta which are related to Alzheimer’s disease pathology.
While the following is a simplified explanation, these are the general steps taken to estimate isotope fractionation using Gaussian. To start, an input file must be built that includes the cartesian coordinates of the atoms in the molecule of interest. This is done in GaussView, Gaussian’s visualization software, by either using templates provided by the software for constructing larger molecules, or building your own manually. When building manually, it’s best to find as much information about the molecule as possible before beginning. This includes information like bond lengths (distance between the centre of atoms, angles between atoms, coordination number (number of atoms bond with the metal atom), and symmetry. Information also required (but which would have made the previous sentence exceedingly lengthy) includes the overall charge of the molecule and information on the molecular electronic state which relates to the spin of the electrons in the molecule. All of this information (and more) is needed in order to optimize the molecular structure. Optimizing the molecular structure means that the total energy of the electrons in the system cannot be lowered by transferring one or more electrons to different orbitals. Once the input file is ready, optimization calculations are performed in Gaussian and can take anywhere from several seconds to several months depending on the complexity of the molecule! Once the structure is optimized, vibrational frequencies for the molecule can be computed. Molecular vibrations occur when atoms in a molecule are in periodic motion while the molecule has constant translational and rotational motion. Generally, a molecule will have 3N-6 vibrational frequencies, where N is the number of atoms present. For example, H2O has 3 vibrational frequencies. I’m interested in the way the vibrational frequencies change depending on which isotope of the central metal atom is present. Therefore, I perform my calculations by specifying in the input file that for example, the zinc atom present is 64Zn. The same calculation is then repeated for 66Zn which will give the molecule slightly different vibrational frequencies than the molecule with 64Zn. The computed frequencies can then be substituted into the equations below to compute lnß values, a measure of isotope fractionation.
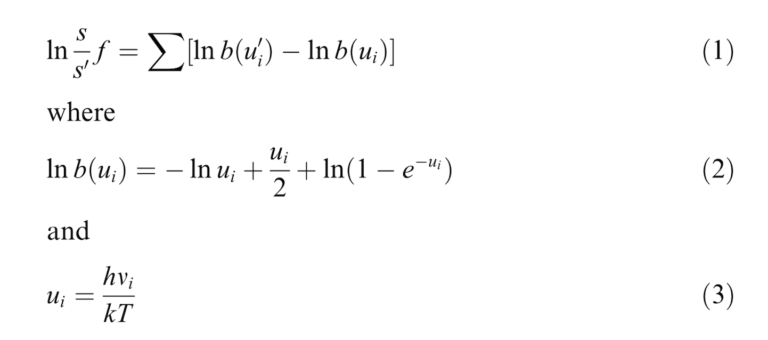
The values aren’t as meaningful on their own, but when compared to those produced for other molecules, you can gain an idea of which molecules would tend to be enriched in heavier isotopes.
While most of this post has been quite heavy in terms of content, I also had some great experiences outside of the office. Dr. Abe made a real effort to make my experience in Japan as fruitful as possible and arranged for me to attend a seminar related to Alzheimer’s disease research at the Tokyo Institute of Technology which was a great opportunity to get exposed to some current research. She also arranged for us to meet with Dr. Yasuhiko Fujii and Dr. Masao Nomura, the researchers who made the first esperimental observations of the nuclear field shift, a mass-independent type of isotope fractionation. This was a real treat for me as anyone who took Dr. Kurt Kyser’s isotope geochemistry course would remember him speaking about this topic. On top of a great lecture given by Dr. Fujii on their research, I also got a tour of their lab which houses a 40-year-old thermal ionization mass spectrometer.

Thermal ionization mass spectrometer. From left to right: Dr. Nomura, myself, Dr. Fujii.
To wrap up our visit with Dr. Fujii and Dr. Nomura, we went to a fantastic izakaya (Japanese-style pub) where we had a great meal that included several courses and a healthy amount of sake. Dr. Fujii has vowed that next time I visit, we’ll go to a karaoke bar. I think I’ll have to take him up on that, but I’ll need to practice first!
I was very much out of my element in the theoretical chemistry department at Tokyo Metropolitan University, but I’m very lucky to have been given the opportunity by the Kimberley Foundation and their Hugh C. Morris Fellowship to work with such talented researchers. I also cannot thank Dr. Abe and her students enough for being such incredible hosts. They took me in under extenuating circumstances and did everything they could to make me feel welcome and make this an amazing learning experience for me. They even included me in their research group photo that they happened to take while I was there.
While Gaussian/GAMESS are powerful tools for theoretical estimation of isotope fractionation, it is most effectively used when coupled with experimental observations. I hope that with an understanding of both the theoretical and experimental sides of isotope geochemistry, I will continue to grow and develop as a researcher, and make meaningful contributions to this field of research.
Now that I’ve returned to Kingston, I’ve got some time to catch up on some of my own lab work before heading off on the next phase of the journey on September 1st at Imperial College London!
Kaj